
Morbus Castleman ist eine sehr seltene Krankheit, unter der verschiedene, klinisch heterogene Krankheitsbilder, mit bestimmten Veränderungen von einem oder mehreren Lymphknoten zusammengefasst werden. Die Ursachen, Diagnosekriterien und Behandlungen unterscheiden sich jedoch je nach Krankheitsbild bzw. Form von Castleman.
Der unizentrische Morbus Castleman (UCD) kann in fast allen Fällen relativ problemlos durch eine Entfernung des betroffenen Lymphknotens geheilt werden. Der multizentrische Morbus Castleman (MCD) ist dagegen eine Systemerkrankung mit einer potenziell lebensbedrohlichen Symptomatik.
Angesichts der Komplexität der Erkrankung erleben viele Betroffene nicht selten lange diagnostische und therapeutische Irrwege. Hinzu kommt, dass Morbus Castleman eine sehr seltene Krankheit ist. In den USA erkranken ca. 4.300 – 5.200 Patienten pro Jahr. In Deutschland wird die Anzahl auf ca. 20-200 Patienten pro Jahr geschätzt. Dies führt dazu, dass viele Ärzte die Krankheit nicht kennen, die Pharmaindustrie wenig Interesse hat, Forschung zu betreiben und Patienten oft alleine dastehen.
Aber ganz so hoffnungslos ist die Situation jedoch nicht. Mit David Fajgenbaum (Arzt und Betroffener) und dem CDCN.org stehen viele hilfreiche Informationen zu Morbus Castleman in englischer Sprache zur Verfügung, werden Gelder gesammelt, Forschung vorangetrieben und eine weltweite Community aufgebaut.
- Die Suche mit deutschen Suchbegriffen führt nicht immer zu den aktuellen und lebensrettenden Informationen. Mit dieser Seite möchte ich eine „Brücke“ zu diesen Inhalten bauen, ergänzt um Links zu Inhalten in deutscher Sprache.
Die verschiedenen Unterformen von Morbus Castleman werden nach der Anzahl der Lymphknoten oder nach der Region der betroffenen Lymphknoten klassifiziert:
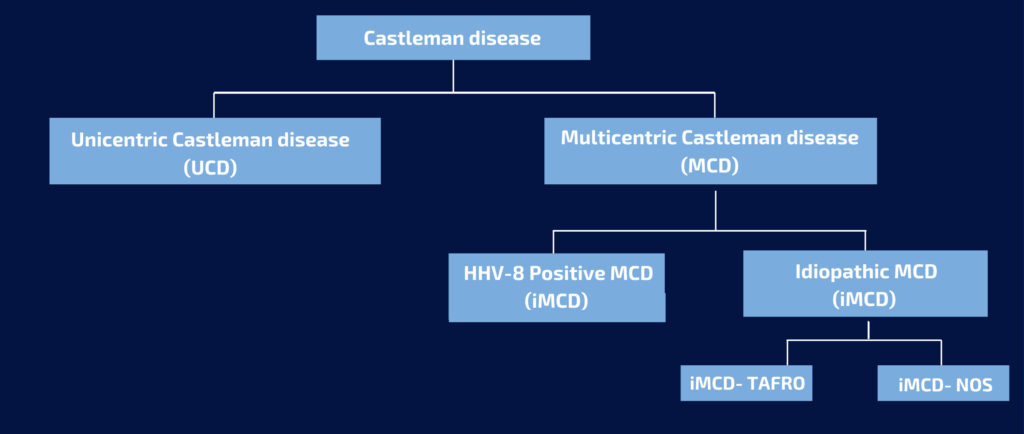
Der unizentrische Morbus Castleman (UCD – Unicentric Castleman Disease) betrifft nur eine Lymphknotengruppe oder einen einzelnen Lymphknoten. Sie ist die häufigste und betrifft 50% der Fälle in der Bevölkerung. Darüber hinaus betrifft sie eher Kinder und junge Erwachsene.
Die multizentrische Castleman-Krankheit (MCD – Multicentric Castleman Disease), die mit der Unterfamilie der Herpesviren (HHV-8) assoziiert ist, betrifft Erwachsene und macht 8-25% der Fälle aus.
Der idiopathische multizentrische Morbus Castleman (iMCD – idiopathic multicentric Castleman Disease) betrifft mehrere Lymphknotenbereiche. Idiopathisch bedeutet, die Ursache ist unbekannt. Sie tritt in jedem Alter auf, aber ältere Erwachsene sind oft am stärksten betroffen. Insgesamt macht sie 25% der Fälle aus. NOS steht für not otherwise specified.
TAFRO
Thrombozytopenie – Blutplättchenmangel
Anasarka – Wassereinlagerung in der Unterhaut
Fibrose – Bindegewebsvernarbung / -verhärtung
Renales – Nieren-Versagen
Organomegalie – Organvergrößerung
Die Krankheit ist sehr selten und dadurch wenig bekannt. Gleichzeitig ist die Diagnose sehr komplex. Durch eine sogenannte Differenzialdiagnose (DD) wird sie von anderen Krankheitsbildern mit ähnlichen Symptomen abgegrenzt. Berücksichtigt werden muss auch die Einnahme von Medikamenten. Vermutlich wird ein beträchtlicher Teil der Castleman-Erkrankung nie korrekt diagnostiziert.
Um Morbus Castleman sicher zu diagnostizieren, ist es extrem wichtig, dass Sie Ihren Arzt darüber informieren, falls Sie von einer der folgenden Erkrankungen betroffen sind:

Kann all dies ausgeschlossen werden, müssen verschiedene Untersuchungen gemacht werden:
- Untersuchung der Lymphknoten: Liegt eine Vergrößerung vor? Wenn ja, wird Ihnen voraussichtlich ein Lymphknoten entnommen, um diesen histologisch zu untersuchen. Dies erfolgt durch einen Pathologen.
- Bildgebung (Ultraschall, CT) u.a. des Bauchraumes, um zu sehen, ob Leber und/oder Milz vergrößert sind.
- Blutuntersuchungen, um verschiedene Blutwerte zu bestimmen.
Nicht alle Patienten mit derselben Unterform weisen die gleichen Symptome auf. Bei einigen treten nur wenige milde Symptome auf, bei anderen kann es zu einem lebensbedrohlichen Organversagen kommen.
Unizentrischer Morbus Castleman – UCD
Symptome UCD
In der Regel haben Patienten mit UCD keine Symptome (asymptomatisch) außer Beschwerden aufgrund des vergrößerten Lymphknotens. Einige Patienten können iMCD-ähnliche Symptome zeigen.; andere können im weiteren Verlauf einen paraneoplastischen Pemphigus entwickeln, der eine sehr ernste Erkrankung ist, die eine schnelle Behandlung erfordert.
Behandlungsmöglichkeiten UCD
Die chirurgische Entfernung des vergrößerten Lymphknotens ist das Mittel der Wahl bei UCD. Wenn eine Entfernung nicht möglich oder kurativ ist, kann auch eine Bestrahlung erfolgen.
Die jeweils aktuellsten Informationen zu den Diagnosekriterien und Behandlungsmöglichkeiten gibt es unter www.cdcn.org Für genaue Informationen zu den einzelnen Therapieoptionen sprechen Sie bitte mit Ihrem Arzt.
Multizentrischer Morbus Castleman – MCD
Symptome MCD (HHV-8-assoziierte MCD, POEMS-MCD, iMCD)
Patienten mit iMCD oder HHV-8-assoziierter MCD können ein breites Spektrum an Symptomen aufweisen, das von leichten grippeähnlichen Episoden bis zu schwerem, lebensbedrohlichem Multiorganversagen reicht.
Einige der systemischen Symptome der MCD umfassen:
• Fieber und Nachtschweiß
• Müdigkeit
• Appetitlosigkeit
• Kachexie (Schwäche und Abmagerung des Körpers, Muskelschwund, ungewollter
Gewichtsverlust)
• Übelkeit und Erbrechen
• Taubheitsgefühl in den Händen und Füßen
• Vergrößerte Leber und/oder Milz
• Leber- und/oder Nierenfunktionsstörungen
o Verminderte Urinausscheidung und systemische Toxizität aufgrund von Nierenversagen
• Flüssigkeitsansammlung (Ödeme, Aszites)
• Veränderungen, die sich im Blutbild zeigen:
o Anämie
o Hypoalbuminämie
o CRP-Anstieg
o zu viel oder zu wenig Blutplättchen
• Bildung von Kirschhämangiomen (kirschrote Papeln, die sich durch ein abnormales Wachstum
von Blutgefäßen auf der Haut bilden)
• Blutergüsse, leichte Blutungen und Infektionsgefahr aufgrund von Knochenmarkversagen
Behandlungsmöglichkeiten
HHV-8-assoziierte MCD
Die Behandlung von HHV-8-assoziierter MCD mit einer B-Zell-Depletionstherapie (z.B. Rituximab) ist hochwirksam. Gelegentlich sind zusätzliche Behandlungen, wie z. B. zytotoxische Chemotherapien, erforderlich. Wenn der Patient HIV-positiv ist, ist eine antiretrovirale Therapie zur Kontrolle der HIV-Infektion sehr wichtig.
iMCD
Die Behandlung sollte zunächst mit einem IL-6-Antikörper erfolgen, in der ersten Wochen zusammen mit Kortison. Das Medikament ist gut verträglich und bei etwa bis zu 50 % der iMCD-Patienten hoch wirksam. Basierend auf dem, was derzeit über das Medikament und die Krankheit bekannt ist, muss das Medikament über Jahre gegeben werden
Für Patienten, bei denen dieses Medikament nicht wirkt, können die folgenden Therapieklassen in Betracht gezogen:
- Konventionelle entzündungshemmende (z. B. Kortikosteroide) und immunsuppressive Therapie: Kortikosteroide können die Symptome während eines akuten iMCD-Schubs verbessern. Nach Absetzen erleiden aber die meisten Patienten einen Rückfall.
- Immunsuppressive Medikamente unterdrücken die Vermehrung und Aktivierung von Immunzellen. Unter anderem auch, weil einige Ärzte die MCD eher wie eine systemische Entzündungserkrankung behandeln.
- Die B-Zell-Depletionstherapie, die bei HHV-8-assoziierter MCD angewendet wird, bietet nur selten eine langfristige Krankheitskontrolle bei iMCD.
- Chemotherapie: Chemotherapieschemata, die traditionell zur Behandlung von Lymphomen eingesetzt werden, haben sich auch bei schwer erkrankten iMCD-Patienten als wirksam erwiesen. Sie eliminieren einen großen Teil der Entzündungszellen. Allerdings treten häufig Rückfälle auf und die Nebenwirkungen sind erheblich.
- Therapien, die auf andere Zytokine und Entzündungswege abzielen: Diese potenziellen Behandlungsoptionen müssen weiter erforscht werden, insbesondere für Patienten, die nicht auf eine Anti-IL-6-Therapie ansprechen.
Was verursacht CD?
Die Daten deuten darauf hin, dass die UCD durch eine unnormale Vermehrung der sogenannten follikulären dendritischen Zellen verursacht wird. Diese Zellen tragen genetische Mutationen, die ihnen Überlebens- und Vermehrungsvorteile verleihen. Dies führt dazu, dass sich der betroffene Lymphknoten vergrößert.
HHV-8-assoziierte MCD wird durch eine Infektion mit HHV-8 (Humanes Herpesvirus-8) verursacht. Bei viele Patienten mit HHV-8-assoziierter MCD ist das Immunsystem nicht mehr intakt – entweder durch eine HIV-Infektion oder eine andere Ursache (z. B. eine genetische Störung, eine immunsuppressive Medikation). Das Virus treibt die Produktion von IL-6 an, die zu den Symptomen der MCD führen.
Über die Ursachen von iMCD ist weit weniger bekannt. Interleukin-6 (IL-6), Botenstoff (Zytokin), der bei Entzündungen eine wichtige Rolle spielt, ist in vielen Fällen von iMCD ein Krankheitstreiber. Die Ursache des erhöhten IL-6 und anderer entzündlicher Zytokine ist unbekannt. Dazu gibt es vier Hypothesen, die derzeit intensiv untersucht werden:
- Autoimmun-Hypothese: Es könnten Antikörper vorhanden sein, die sich gegen gesundes Gewebe der Patienten richten und das Immunsystem zu einer Überaktivität anregen, wodurch Entzündungsmoleküle (Zytokine) freigesetzt werden
- Autoinflammatorische Hypothese: Möglicherweise besteht ein genetischer Defekt, durch den das Immunsystem abgeschaltet werden kann, was wiederum zur Überproduktion führt.
- Hypothese des paraneoplastischen Syndroms: Es können erworbene onkogene („krebsverursachende“) Mutationen vorliegen, die bewirken, dass Zytokine entweder von Zellen innerhalb der CD-Lymphknoten oder von Zellen, die mit einer gleichzeitig vorhandenen Krebserkrankung assoziiert sind, ausgeschüttet werden.
- Pathogen-Hypothese: Vielleicht gibt es einen unentdeckten Erreger (d. h. ein anderes Virus als HHV-8), der eine unkontrollierte Immunaktivierung und Zytokinausschüttung verursacht, ähnlich wie HHV-8 es bei HHV-8-assoziierter MCD tut.
Was sind die Risikofaktoren für die Entwicklung von CD?
Es ist sehr wenig über die Risikofaktoren für diese Erkrankung bekannt. Bislang wurden keine Risikofaktoren für UCD oder iMCD identifiziert, obwohl die Möglichkeit einer genetischen Prädisposition untersucht wird. Zu den Risikofaktoren für eine HHV-8-assoziierte MCD gehören alle Faktoren, die das Immunsystem signifikant unterdrücken und somit Personen für eine unkontrollierte HHV-8-Infektion prädisponieren (HIV, genetische Störungen, immunsuppressive Medikamente und Organtransplantationen).
Können UCD und MCD verwechselt werden?
Beim UDC gibt seltene Fälle, die ein Rezidiv in der gleichen oder einer anderen Region entwickeln. Es kann herausfordernd sein, zu unterscheiden, ob wirklich ein UCD-Rezidiv vorliegt oder der UCD tatsächlich ein MCD ist.
Ist CD eine Form von Krebs?
Die Daten deuten darauf hin, dass es sich bei UCD wahrscheinlich um einen neoplastischen Prozess handelt, der durch eine abnorme Proliferation von follikulären dendritischen Zellen verursacht wird. Es wurde festgestellt, dass diese Zellen Mutationen besitzen, die ihnen Proliferations- und Überlebensvorteile verleihen. Patienten mit HHV-8-assoziierter MCD und iMCD haben manchmal Anzeichen und Symptome, die schwer von einem aggressiven Lymphom zu unterscheiden sind. Es gibt auch Fallberichte, bei denen anschließend ein hämatologisches Malignom diagnostiziert wurde. Stichwort Autoimmun.
Wie ist der Zusammenhang zwischen HHV-8-negativer/idiopathischer MCD und dem Krebsrisiko?
Hämatologische Malignome werden innerhalb von zwei Jahren nach der Diagnose einer iMCD mit erhöhter Häufigkeit beobachtet.Der genaue Grund für dieses erhöhte Risiko ist nicht bekannt.
Es wird empfohlen, allen neu diagnostizierten HHV-8-negativen iMCD-Patienten auf ein zugrunde liegendes Malignom zu untersuchen.
Welche abnormalen Laborwerte werden typischerweise bei CD gesehen?
- Die meisten UCD Patienten haben keine abnormalen Blutwerte, aber bei einigen können ähnliche Veränderungen wie bei iMCD (unten) auftreten.
- Patienten mit iMCD und HHV-8-assoziierte MCD weisen häufig mehrere abnorme Blutwerte auf.
- iMCD Patienten mit dem TAFRO-Subtyp können zusätzlich zu typischen iMCD-Symptomen eine Thrombozytopenie, Anasarca, Fibrose, Nierenversagen (Renal) und/oder eine vergrößerte Leber und/oder Milz (Organanomegalie) aufweisen.
- Andere iMCD Patienten haben oft eine erhöhte Thrombozytenzahl, weniger schwere Flüssigkeitsansammlungen und erhöhte Gammaglobulinwerte.
Welche Lymphknotenveränderungen werden bei CD gesehen?
CD-Lymphknoten zeigen unter dem Mikroskop mehrere Merkmale, die in eine von drei Varianten eingeteilt werden können:
- Hyalin-vaskulär oder hypervaskulär (HV): gekennzeichnet durch verbreiterte Mantelzonen, die aus konzentrischen Ringen kleiner Lymphozyten in einem „Zwiebelschalen“-Muster um kleine atrophische Keimzentren bestehen, Kapselfibrose mit breiten fibrösen Bändern im gesamten Lymphknoten, eine erhöhte Anzahl lymphatischer Follikel, durchdringende hyalinisierte Gefäße und dysplastische follikuläre dendritische Zellen (FDCs). Wird am häufigsten bei UCD gesehen.
- Plasmazellen (PC): Die Keimzentren sind eher hyperplastisch als atrophisch, die interfollikuläre Region enthält Platten von Plasmazellen und Gefäßproliferationen, das FDC-Netzwerk ist normal und die Lymphknotenarchitektur ist erhalten.
- Die Bedeutung dieser histopathologischen Varianten ist unklar, da es Berichte über Übergänge zwischen HV- und PC-Varianten bei nachfolgenden Biopsien sowie über das gleichzeitige Vorhandensein beider Typen in getrennten Lymphknoten derselben Patientin / desselben Patienten gibt. Außerdem können dieselben Merkmale auch bei anderen Krankheiten auftreten, die sich ähnlich wie CD verhalten.
Wie wird sich dies auf meine Lebenserwartung auswirken?
Es ist unmöglich zu wissen, wie sich diese Diagnose auf Ihre Lebenserwartung auswirkt, aber basierend auf großen Studien mit Hunderten von Patienten können die folgenden Verallgemeinerungen gemacht werden:
- UCD hat normalerweise keinen Einfluss auf die Lebenserwartung (>95% überleben mindestens 5 Jahre nach der Diagnose). Es gibt jedoch UCD-Patienten, die einen paraneoplastischen Pemphigus, Lymphome oder Sarkome entwickeln, die tödlich verlaufen können.
- HHV-8-assoziierte MCD: Ungefähr 90 % der überleben mindestens 5 Jahre nach der Diagnose, wenn sie angemessen mit einer B-Zell-Depletionstherapie behandelt werden.
- iMCD: Etwa 55-75 % der Patienten überleben mindestens 5 Jahre nach der Diagnose. Mit dem Aufkommen von spezifischen Anti-IL-6-Therapien ist eine Verbesserung der Überlebensraten zu erwarten.
Ist es sicher für mich, schwanger zu werden?
Bislang gibt es keine Studien zu dieser wichtigen Frage, daher können wir keine endgültige Antwort geben.
